
bioBakery Workflows
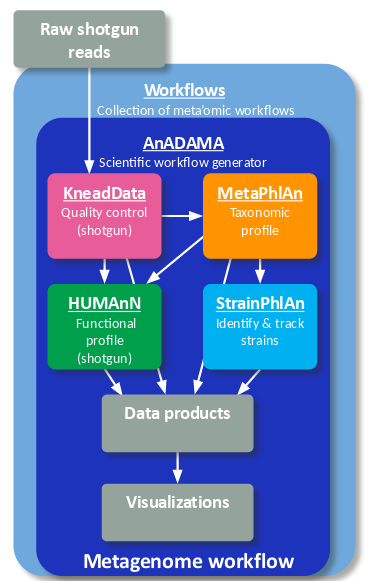
bioBakery workflows is a collection of workflows and tasks for executing common microbial community analyses using standardized, validated tools and parameters. Quality control and statistical summary reports are automatically generated for most data types, which include 16S amplicons, metagenomes, and metatranscriptomes. Workflows are run directly from the command line and tasks can be imported to create your own custom workflows. The workflows and tasks are built with AnADAMA2 which allows for parallel task execution locally and in a grid compute environment.
For more information on the technical aspects:
User Manual || User Tutorial || Forum
Features
bioBakery workflows inherit many features from AnADAMA2 (indicated by *).
- Reproducible workflows
- Full reports including visualizations are generated for each workflow which include all commands run and software versions
- Efficient workflows
- Simple user interface
- A single command runs a complete workflow which includes quality control, taxonomic and functional profiling
- Local parallelization*
- Grid meta-scheduler (SLURM and SGE)*
- Jobs are dispatched, monitored, and logged
- Jobs exceeding time or memory estimates are resubmitted
- Simple user interface
- Reproducible workflows
Requirements
- Python (version >= 2.7)
- Operating system (Linux or Mac)
- AnADAMA2 (installed automatically)
- Workflow dependencies (see the bioBakery workflows User Manual for a list of the dependencies for each workflow (i.e. KneadData, MetaPhlAn2, etc))
Getting started
Installation
bioBakery workflows can be installed with pip, conda, or Docker.
Install with Pip
$ pip install biobakery_workflows
- Installing with pip will only install the core software and dependencies. It will not install the dependencies for individual workflows (ie Kneaddata, MetaPhlAn, etc).
- If you do not have write permissions to
/usr/lib/, then add the option--userto the install command. This will install the python package into subdirectories of$HOME/.localon Linux.
Install with Docker
$ docker run -it biobakery/workflows bash
- The image will include all dependencies for all workflows (ie Kneaddata, MetaPhlan, etc.) excluding those dependencies that have licenses (e.g. USEARCH).
How to run
Basic usage
Whole metagenome shotgun sequencing data can be processed through read-level quality control (KneadData), taxonomic profiling (MetaPhlAn), functional profiling (HUMAnN), and strain profiling (StrainPhlAn) to generate a report with publication-ready figures with two workflow commands.
$ biobakery_workflows wmgx --input $INPUT --output $OUTPUT_DATA
$ biobakery_workflows wmgx_vis --input $OUTPUT_DATA --output $OUTPUT_VIS --project-name $PROJECT
$INPUT: A directory containing shotgun sequencing data (i.e. fasta/fastq in gzipped format)$OUTPUT_DATA: A directory to write the data products (i.e. abundance tables)- This folder is the output folder for the first command and the input folder for the second command
$OUTPUT_VIS: A directory to write the visualization products (i.e. report, figures, data tables)$PROJECT: The name of the project (included in the report title page)- Add the options
--local-jobs 8 --threads 4to run8local jobs at a time each with4threads. - Add the option
--grid-jobs 100to run100grid jobs at a time.