
HUMAnN 2.0 is a pipeline for efficiently and accurately profiling the presence/absence and abundance of microbial pathways in a community from metagenomic or metatranscriptomic sequencing data (typically millions of short DNA/RNA reads). This process, referred to as functional profiling, aims to describe the metabolic potential of a microbial community and its members. More generally, functional profiling answers the question “What are the microbes in my community-of-interest doing (or capable of doing)?”
For more information on the technical aspects of HUMAnN 2.0:
User manual || Tutorial || Forum
Citation:
Franzosa EA*, McIver LJ*, Rahnavard G, Thompson LR, Schirmer M, Weingart G, Schwarzberg Lipson K, Knight R, Caporaso JG, Segata N, Huttenhower C. Species-level functional profiling of metagenomes and metatranscriptomes. Nat Methods 15: 962-968 (2018).
Interested in trying out the latest version? We're in the late-stage testing for a new version HUMAnN 3.0
- Community functional profiles stratified by known and unclassified organisms
- MetaPhlAn2 and ChocoPhlAn pangenome database are used to facilitate fast, accurate, and organism-specific functional profiling
- Organisms included are Archaea, Bacteria, Eukaryotes, and Viruses
- Considerably expanded databases of genomes, genes, and pathways
- A simple user interface (single command-driven flow)
- The user only needs to provide a quality-controlled metagenome or metatranscriptome
- Accelerated mapping of reads to reference databases (including run-time generated databases tailored to the input)
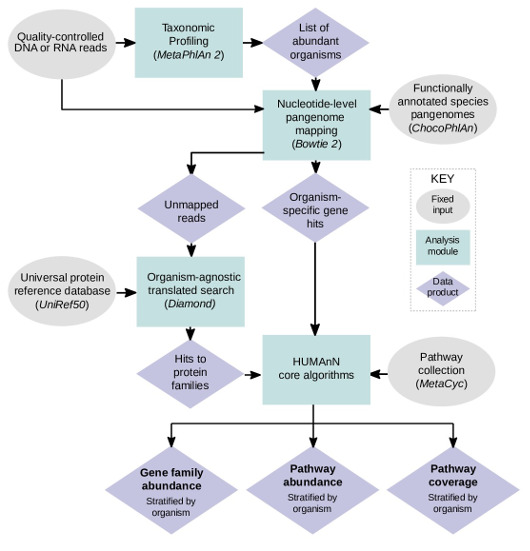
- MetaPhlAn 2.0
- Bowtie2 (version >= 2.1) (see NOTE)
- Diamond (0.9.0 > version >= 0.8.22) (see NOTE)
- MinPath (see NOTE)
- Python (version >= 2.7)
- Memory (>= 16 GB)
- Disk space (>= 10 GB [to accommodate comprehensive sequence databases])
- Operating system (Linux or Mac)
NOTE: Bowtie2, Diamond, and MinPath are automatically installed when installing HUMAnN 2.0. If these dependencies do not appear to be installed after installing HUMAnN 2.0 with pip, it might be that your environment is setup to use wheels instead of installing from source. HUMAnN 2.0 must be installed from source for it to also be able to install dependencies. To force pip to install HUMAnN 2.0 from source add one of the following options to your install command, “–no-use-wheel” or “–no-binary :all:”.
When installing HUMAnN 2.0, please also download and install MetaPhlAn 2.0. You can then add the MetaPhlAn2 folder to your $PATH or you can provide its location when running HUMAnN 2.0 with the option “–metaphlan $DIR” (replacing $DIR with the full path to the MetaPhlAn2 folder). Bowtie2, Diamond, and Minpath will be automatically installed when you install HUMAnN 2.0
-
- Install HUMAnN 2.0
- $ pip install humann2
- This command will automatically install MinPath (and a new version of glpk) along with Bowtie2 and Diamond (if they are not already installed).
- To bypass the install of Bowtie2 and Diamond, add the option “–install-option=’–bypass-dependencies-install'” to the install command.
- To build Diamond from source during the install, add the option “–install-option=’–build-diamond'” to the install command.
- To overwite existing installs of Bowtie2 and Diamond, add the option “–install-option=’–replace-dependencies-install'” to the install command.
- If you do not have write permissions to ‘/usr/lib/’, then add the option “–user” to the HUMAnN 2.0 install command. This will install the python package into subdirectories of ‘~/.local’ on Linux. Please note when using the “–user” install option on some platforms, you might need to add ‘~/.local/bin/’ to your $PATH as it might not be included by default. You will know if it needs to be added if you see the following message humann2: command not found when trying to run HUMAnN2 after installing with the “–user” option.
- Test the HUMAnN2 install (Optional)
- $ humann2_test
- To also run tool tests, add the option “–run-functional-tests-tools”.
- To also run end-to-end tests, add the option “-run-functional-tests-end-to-end”. Please note these tests take about 20 minutes to run. Also they require all dependencies of HUMAnN 2.0 be installed in your PATH.
- Try out a HUMAnN 2.0 demo run (Optional)
- Download the HUMAnN 2.0 source with demos: humann2.tar.gz
- $ tar zxvf humann2.tar.gz
- $ humann2 –input humann2/examples/demo.fastq –output $OUTPUT_DIR
- When running this command, $OUTPUT_DIR should be replaced with the full path to the directory you have selected to write the output from the HUMAnN 2.0 demo run.
- Other types of demo files are included in this folder and can be run with the exact same command.
- Demo ChocoPhlAn and UniRef databases are also included in the download. The demo ChocoPhlAn database is located a humann2/data/chocophlan_DEMO and the demo UniRef database is located a humann2/data/uniref_DEMO. Until the full databases are downloaded HUMAnN 2.0 will run with the demo database by default.
- Download the ChocoPhlAn database (approx. size = 5.6 GB)
- $ humann2_databases –download chocophlan full $DIR
- When running this command, $DIR should be replaced with the full path to the directory you have selected to store the database.
- This command will update the HUMAnN2 configuration file, storing the location you have selected for the ChocoPhlAn database. If you move this database and would like to change the configuration file, please see the Configuration Section of the HUMAnN 2.0 User Manual. Alternatively, if you move this database, you can provide the location by adding the option “–nucleotide-database $DIR” when running HUMAnN 2.0.
- Download a UniRef database (only download one database: UniRef50 full, UniRef50 EC filtered, UniRef90 full, or UniRef90 EC filtered)
- Download one of the following databases (replacing $DIR with the location to store the database):
- To download the UniRef90 EC filtered database (RECOMMENDED, approx. size = 846 MB):
- $ humann2_databases –download uniref uniref90_ec_filtered_diamond $DIR
- To download the full UniRef90 database (approx. size = 11 GB):
- $ humann2_databases –download uniref uniref90_diamond $DIR
- To download the UniRef50 EC filtered database (approx. size = 239 MB):
- $ humann2_databases –download uniref uniref50_ec_filtered_diamond $DIR
- To download the full UniRef50 database (approx. size = 4.6 GB):
- $ humann2_databases –download uniref uniref50_diamond $DIR
- To download the UniRef90 EC filtered database (RECOMMENDED, approx. size = 846 MB):
- Select a full database if you are interested in identifying uncharacterized proteins in your data set. Alternatively, select an EC filtered database if you have limited disk space and/or memory. For example, a run with 13 million reads (approximately 7 GB fastq file) passed as input to the translated search step, using a single core, ran in about 4 hours with a maximum of 6 GB of memory using the UniRef50 EC filtered database. The same input file using the UniRef50 full database ran in 25 hours, with a single core, with a maximum of 11 GB of memory.
- The download command will update the HUMAnN2 configuration file, storing the location you have selected for the UniRef database. If you move this database and would like to change the configuration file, please see the Configuration Section of the HUMAnN2 User Manual. Alternatively, if you move this database, you can provide the location by adding the option “–protein-database $DIR” when running HUMAnN 2.0.
- Download one of the following databases (replacing $DIR with the location to store the database):
- Download the utility scripts mapping files database (approx. size = 0.5 GB) (Optional)
- $ humann2_databases –download utility_mapping full $DIR
- When running this command, $DIR should be replaced with the full path to the directory you have selected to store the database.
- Downloading and installing these mapping files will give you additional options when running the humann2_rename_table and humann2_regroup_table utility scripts.
- Install HUMAnN 2.0
$ humann2 –input $SAMPLE –output $OUTPUT_DIR
$SAMPLE = a single file that is one of the following types:
- filtered shotgun sequencing metagenome file (fastq, fastq.gz, fasta, or fasta.gz format)
- mapping results file (sam, bam or blastm8 format)
- gene table file (tsv or biom format)
$OUTPUT_DIR = the output directory
Three output files will be created:
- $OUTPUT_DIR/$SAMPLENAME_genefamilies.tsv*
- $OUTPUT_DIR/$SAMPLENAME_pathcoverage.tsv
- $OUTPUT_DIR/$SAMPLENAME_pathabundance.tsv
where $SAMPLENAME is the basename of $SAMPLE
*The gene families file will not be created if the input file type is a gene table.
Intermediate temp files will also be created:
- $DIR/$SAMPLENAME_bowtie2_aligned.sam
- the full alignment output from bowtie2
- $DIR/$SAMPLENAME_bowtie2_aligned.tsv
- only the reduced aligned data from the bowtie2 output
- $DIR/$SAMPLENAME_bowtie2_index*
- bowtie2 index files created from the custom chochophlan database
- $DIR/$SAMPLENAME_bowtie2_unaligned.fa
- a fasta file of unaligned reads after the bowtie2 step
- $DIR/$SAMPLENAME_custom_chocophlan_database.ffn
- a custom chocophlan database of fasta sequences
- $DIR/$SAMPLENAME_metaphlan_bowtie2.txt
- the bowtie2 output from metaphlan
- $DIR/$SAMPLENAME_metaphlan_bugs_list.tsv
- the bugs list output from metaphlan
- $DIR/$SAMPLENAME_$TRANSLATEDALIGN_aligned.tsv
- the alignment results from the translated alignment step
- $DIR/$SAMPLENAME_$TRANSLATEDALIGN_unaligned.fa
- a fasta file of unaligned reads after the translated alignment step
- $DIR/$SAMPLENAME.log
- a log of the run
- $DIR=$OUTPUT_DIR/$SAMPLENAME_humann2_temp/
- $SAMPLENAME is the basename of the fastq/fasta input file
- $TRANSLATEDALIGN is the translated alignment software selected (rapsearch2 or usearch)
NOTE: $SAMPLENAME can be set by the user with the option “–output-basename <$NEWNAME>”.
The standard workflow involves running HUMAnN2 on each filtered shotgun sequencing metagenome file, normalizing, and then merging output files.
Synthetic datasets from the HUMAnN 2.0 paper:
The following archives include the synthetic datasets that were used in the evaluations described in the paper.