
PARATHAA(Preserving and Assimilating Region-specific Ambiguities in Taxonomic Hierarchical Assignments for Amplicons) is a tool used for the taxonomic assignment of 16S rRNA gene sequences that takes into account the uncertainty associated with using specific variable regions/primers. PARATHAA does this by generating new primer-trimmed phylogenetic trees from reference 16S rRNA gene datasets and then determines the optimal phylogenetic distances within that tree for taxonomic labeling. PARATHAA then can use this tree to assign taxonomy to query 16S rRNA gene sequences by aligning and placing those sequences into the new primer-trimmed reference database.
User manual || Tutorial || Forum
Citation:
Workflow to generate results for Parathaa manuscript: https://github.com/biobakery/manuscript_parathaa
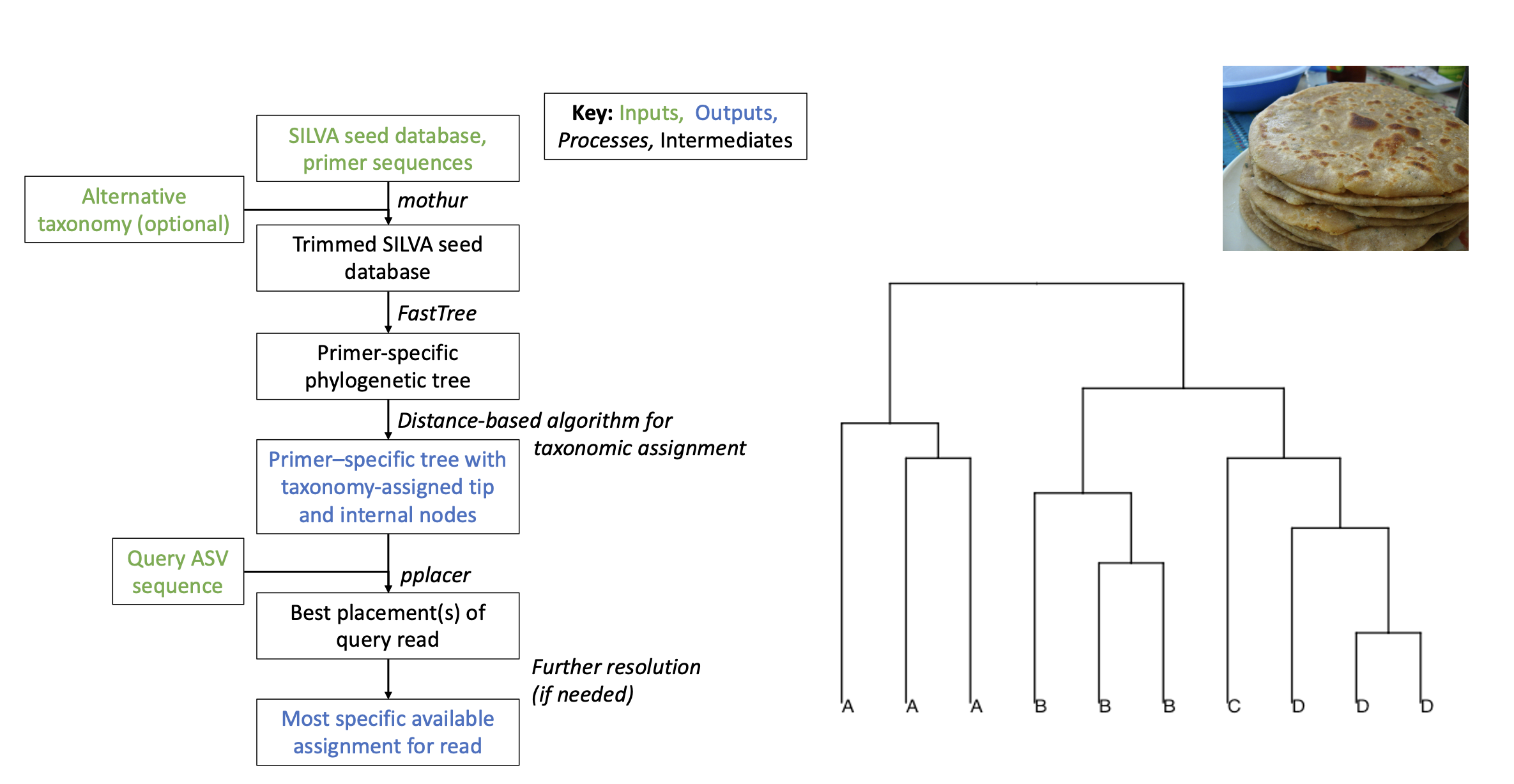
The first step in PARATHAA is to generate a phylogenetic tree of primer-trimmed sequences with taxonomically-labeled internal nodes. PARATHAA accomplishes this in the following way:
- Takes an input 16S rRNA gene reference alignment set and trims those alignments to only the regions amplified by the given primer set.
- Uses the newly trimmed sequences to generate a phylogenetic tree using FastTree.
- Determines the optimal phylogenetic distance cutoffs for each taxonomic group based on the original reference taxonomy.
- This is a key step in the PARATHAA workflow and is accomplished by following:
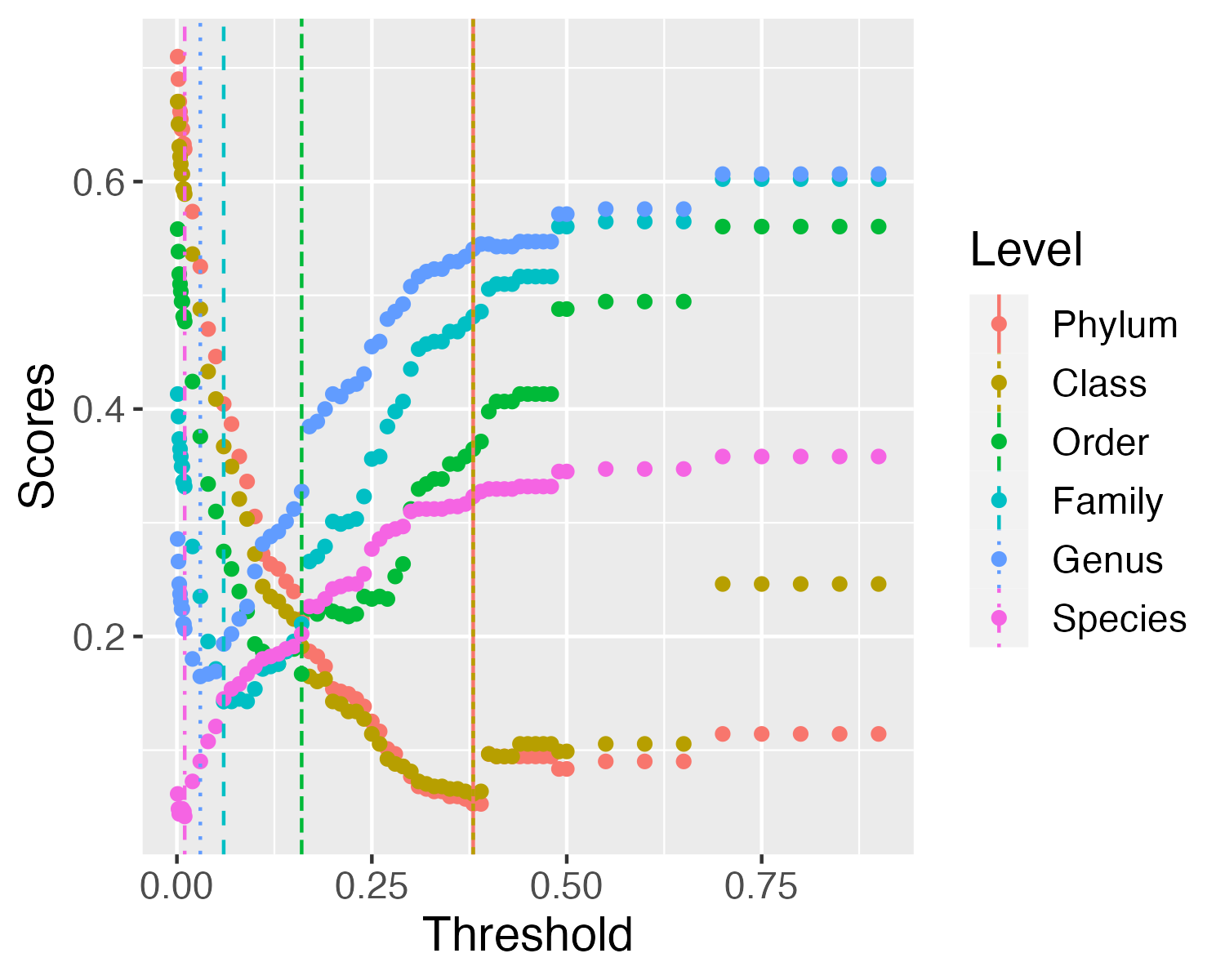
Optimal cophenetic distance thresholds based on the Jukes-Cantor model of nucleotide evolution (as implemented in FastTree) are identified, which will define taxa at each level. For instance, any node whose underlying tips have pairwise distances less than the genus-defining threshold are considered to be of the same genus. To find the optimal thresholds at each taxonomic level, a threshold-finding step tries a range of distance cutoffs for each taxonomic level, and calculates an associated error based on misclassification of sequences (whose taxonomy is known). The chosen threshold is that which minimizes (1) grouping leaves of multiple taxa under a taxon-defining node (“over-merging”), and (2) splitting of sequences from the same taxonomic group into multiple taxon-defining nodes (“over-splitting”).
- Uses these optimal phylogenetic distances to relabel the internal nodes of the trimmed phylogenetic tree.
All of these steps can be completed using the run_tree_analysis.py command with the proper inputs.
The second step of PARATHAA uses the phylogenetic tree, multiple sequence alignment (MSA) and tree data files to create taxonomic assignments to 16S rRNA gene sequences. This step is running using the following command:
run_taxa_assignment.py
PARATHAA will come with a number of pre-computed-trees for this step so that users do not need to generate their own primer trimmed trees for commonly used reference databases. However this is still under development and currently the only pre-computed-tree available is for silva_v138 using V4V5 primers.
Briefly this step takes in the newly created primer trimmed tree, MSA, tree reference files, and thresholding information to assign taxonomy to query 16S sequences by the following steps.
- Aligns query sequences to primer trimmed MSA generated in step 1
- Places those sequences into the primer trimmed tree generated in step 1 using pplacer
- Assigns taxonomy to those query sequences based on their placement and their distance from taxonomically labelled interior nodes. Note the thresholding distance computed in step one determines the apprioriate distance away a node can be to be assigned to a taxonomic level.