
BAQLaVa (Bioinformatic Application for Quantification and Labeling of Viral taxonomy) is a forthcoming bioBakery method for accurate and efficient identification and quantification of known and novel viral taxa from shotgun metagenomic and metatranscriptomic sequencing data.
User manual || Tutorial || Forum
Citation:
Jordan S. L. Jensen, Sagun Maharjan, Philipp C. Münch, Jiaxian Shen, Bailey Bowcutt, Jack T. Sumner, Moreno Zolfo, Claudia Mengoni, Xochitl C. Morgan, Kelsey N. Thompson, Nicola Segata, Long H. Nguyen, Eric A. Franzosa, Curtis Huttenhower
Enhanced multi-omic viral profiling from microbial community sequencing with BAQLaVa
BAQLaVa begins by combining putative viral sequences from RefSeq and ICTV with the Gut Virome Database, Gut Phage Database, and our own Viral Sequence Contigs. These are clustered into a collection of non-redundant “viral genome bins” (VGBs) based on MIUViG standards in a process analogous to our bacterial species-level genome bins (SGBs). BAQLaVa subjects VGBs to nucleotide-level markerization and mapping using algorithms adapted from MetaPhlAn. Then, to boost sensitivity to sequence variation within known viral clades, BAQLaVa incorporates the HUMAnN approach to “tiered search,” aligning unmapped MGX/MTX reads to a broader viral protein database using translated search. Finally, to profile novel viruses, BAQLaVa incorporates deep-learning classifiers based on the GenomeNet-Architect framework and trained on its own VGBs plus existing negative examples from bioBakery databases of non-viral sequences.
Figure 1. Overview of the BAQLaVa workflow for virome profiling and model evaluation. A) BAQLaVa upstream “indexing” includes clustering of viral genome bins (“VGBs”) from input viral sequences and downstream VGB markerization (to identify unique sequences for profiling), ORF calling (for amino acid sequences), and neural network training (for classification). B) Overview of BAQLaVa’s integrated workflow for viral profiling of MGX/MTX through a combination of tiered real-level mapping, assembly and contig classification, and profile harmonization. C) Synthetic evaluation of two key nucleotide-level search parameters to be used in BAQLaVa, per-sequence coverage and total sequence length, as evaluated by recall and precision. D) The same parameters tuned for BAQLaVa’s translated search tier. E) Evaluation of parameter-optimized nucleotide search recall on synthetic viromes of differing composition. F) The same synthetic viromes applied for precision estimation.
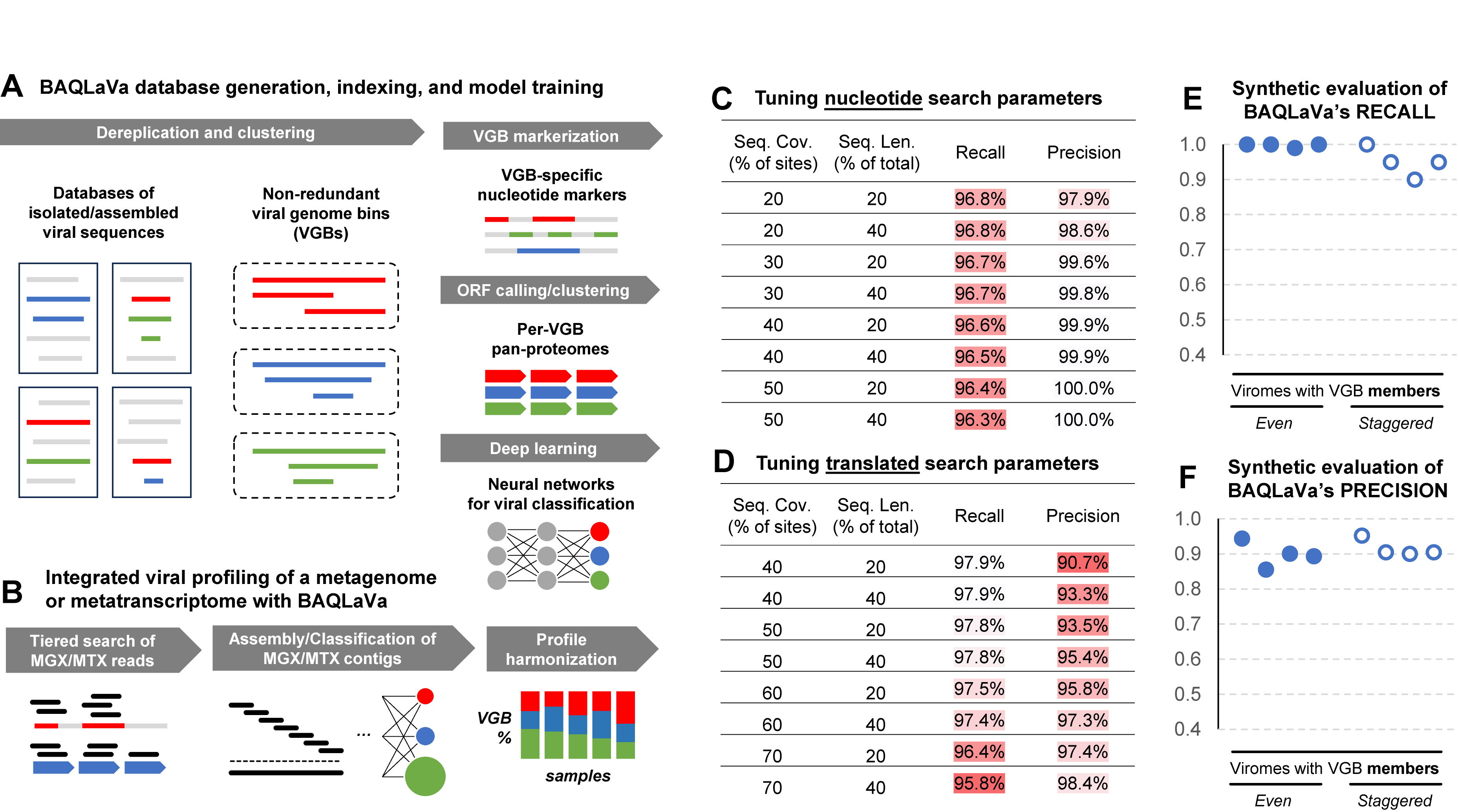
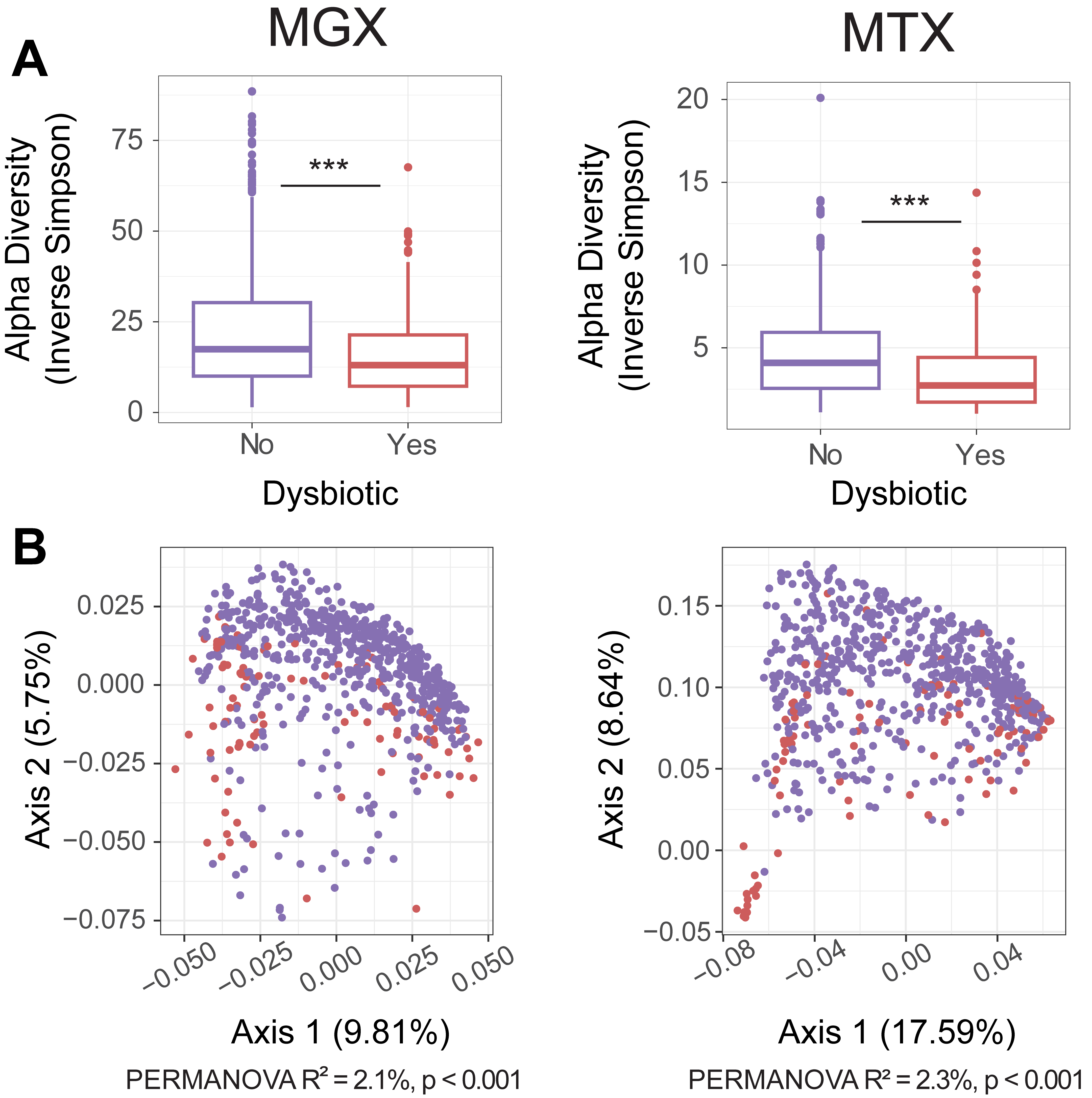
Figure 2. Changes in the human gut virome coincident with Inflammatory Bowel Disease (IBD)-associated dysbiosis. We applied BAQLaVa to metagenomic (MGX) and metatransciptomic (MTX) sequencing from the 1,785 longitudinally collected stool samples from the HMP2 IBDMDB cohort (data from https://ibdmdb.org/). BAQLaVa identified 693 prevalent and abundant VGBs from MGX samples and 192 from MTX samples. Similar to bacterial taxonomic composition during IBD-associated dysbiosis, A) viral species alpha diversity was significantly depleted in dysbiotic versus non-dysbiotic samples (both MGX and MTX) and B) there was a small but significant difference in between-sample (beta) diversity (Bray-Curtis distance) across the two phenotypes as determined by PERMANOVA.
Installation
Install using PyPi:
pip install baqlava
Install using github:
git clone https://github.com/biobakery/baqlava.git
cd baqlava/
pip install .
Note: Please use --user flag if you do not have permission to install the tools as a root user.
pip install baqlava --userDatabase requirements
Usage: (BAQLaVa DB instructions will be shown at the end of the setup completion as a banner message)
baqlava_database --download database baqlava-db /path/to/install_database
BAQLaVa requires two input databases:
- a bowtie2-formatted nucleotide sequence database and
- a DIAMOND-formatted protein sequence database. The above command will download and install the required BAQLaVa DB for the run in the specified path.
Test your installation
baqlava -i <FILE> -o <OUTPUT_DIRECTORY>
When running BAQLaVa v0.5, you have the following options:
--bypass-bacterial-depletion: run your sample through viral profiling without removing bacterial reads first
--bypass-nucleotide-search: run only the translated search component of BAQLaVa
--bypass-translated-search: run only the nucleotide search component of BAQLaVa
--taxonomic-profile: If you sample has previously been profiled with MetaPhlAn, you can speed up the bacterial depletion step and provide a MetaPhlAn taxonomic profile to be used directly