
PICRUSt2 (Phylogenetic Investigation of Communities by Reconstruction of Unobserved States) is a software for predicting functional abundances based only on marker gene sequences. Check out the paper here.
"Function" usually refers to gene families such as KEGG orthologs and Enzyme Classification numbers, but predictions can be made for any arbitrary trait. Similarly, predictions are typically based on 16S rRNA gene sequencing data, but other marker genes can also be used.
User manual || Tutorial || Forum
Citation:
Douglas, G.M., Maffei, V.J., Zaneveld, J.R. et al. PICRUSt2 for prediction of metagenome functions. Nat Biotechnol 38, 685–688 (2020). https://doi.org/10.1038/s41587-020-0548-6
However, if you use PICRUSt2 you also need to cite the below tools.
For phylogenetic placement of reads:
-
EPA-NG (paper, website) - Default placement option.
-
gappa (paper, website)
-
SEPP (paper, website) - If alternative placement option used.
For hidden state prediction:
For pathway inference:
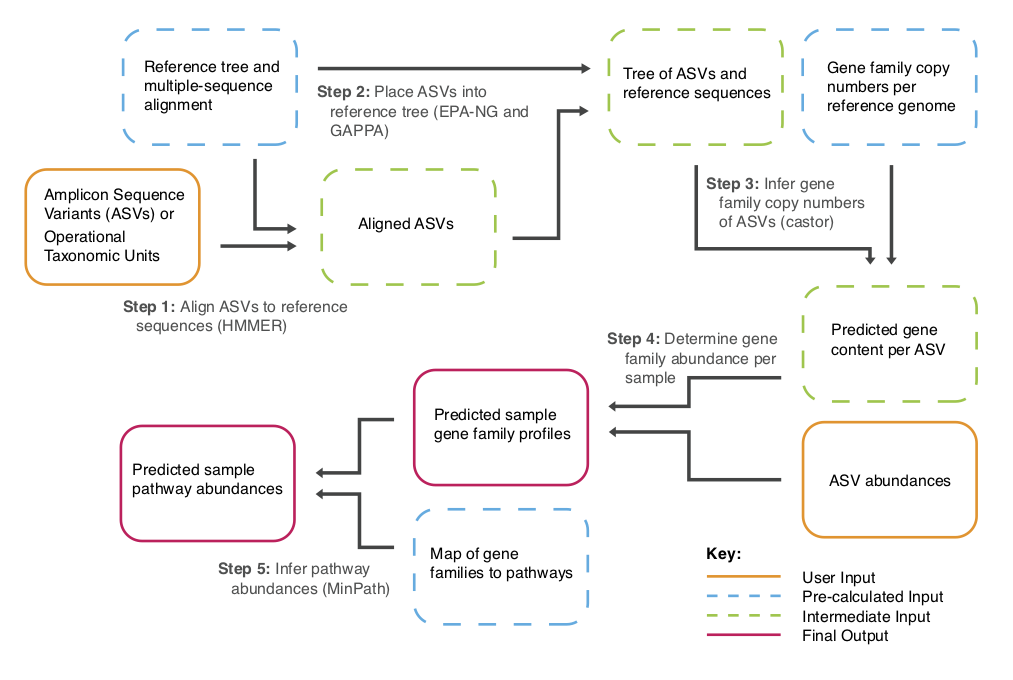
PICRUSt2 includes these and other improvements over the original version:
- Allow users to predict functions for any 16S sequences. Representative sequences from OTUs or amplicon sequence variants (e.g. DADA2 and deblur output) can be used as input by taking a sequence placement approach
- Database of reference genomes used for prediction has been expanded by >10X.
- Addition of hidden-state prediction algorithms from the
castorR package. - Allows output of MetaCyc ontology predictions that will be comparable with common shotgun metagenomics outputs.
- Inference of pathway abundances now relies on MinPath, which makes these predictions more stringent.
Create a new environment with PICRUSt2 installed and activate the environment. If you receive an error about the default reference files missing when you try to run the program you will need to install from source instead.
conda create -n picrust2 -c bioconda -c conda-forge picrust2=2.4.1
conda activate picrust2Download the source tarball, untar, and move into the directory. You can check that you are downloading the latest release here: https://github.com/picrust/picrust2/releases
wget https://github.com/picrust/picrust2/archive/v2.4.1.tar.gz
tar xvzf v2.4.1.tar.gz
cd picrust2-2.4.1/
Create and activate the environment (with requirements) and then install PICRUSt2 with pip.
conda env create -f picrust2-env.yaml
conda activate picrust2
pip install --editable .
Run the tests to verify the installation (note that this wont work if you followed the bioconda install instructions):
pytest
Below is an overview of the PICRUSt2 workflow, which includes example commands for processing 16S sequencing data and getting E.C. number and KEGG ortholog (KO) abundances. The E.C. numbers can then be used to calculate MetaCyc pathway abundances and coverages. Note that there are other gene family databases supported which may be more informative (but which cannot be collapsed to pathways by default). See the side-bar for more details on individual commands.
Note that you can type the option -h to get a description of each below script.
The entire pipeline can be run with this command (details):
picrust2_pipeline.py -s study_seqs.fna -i study_seqs.biom -o picrust2_out_pipeline -p 1
If you would like to run each step individually you can also do that using the below commands. Using these commands is useful when you're running into problems using picrust2_pipeline.py and want to isolate an issue or if you only want to re-run part of the PICRUSt2 pipeline.