
Lateral gene transfer (LGT) is an important mechanism for genome diversification in microbial communities, including the human microbiome. While methods exist to identify LGTs from sequenced isolate genomes, identifying LGTs from community metagenomes remains an open problem. To address this, we developed WAAFLE: a Workflow to Annotate Assemblies and Find LGT Events.
User Manual || User Tutorial || Forum
Citation:
Profiling lateral gene transfer events in the human microbiome using WAAFLE
DOI: 10.1038/s41564-024-01881-w
1 Harvard T.H. Chan School of Public Health, Boston, MA, USA.
2 Massachusetts General Hospital and Harvard Medical School, Boston, MA, USA.
3 Faculty of Computer Science, Dalhousie University, Halifax, Nova Scotia, Canada.
4 The Broad Institute of MIT and Harvard, Cambridge, MA, USA.
5 Department of Pharmacology, Dalhousie University, Halifax, Nova Scotia, Canada.
6 Harvard T.H. Chan School of Public Health, Boston, MA, USA. lnguyen24@mgh.harvard.edu.
7 Massachusetts General Hospital and Harvard Medical School, Boston, MA, USA. lnguyen24@mgh.harvard.edu.
8 Harvard T.H. Chan School of Public Health, Boston, MA, USA. franzosa@hsph.harvard.edu.
9 The Broad Institute of MIT and Harvard, Cambridge, MA, USA. franzosa@hsph.harvard.edu.
#Contributed equally.
-
- Compatibility with SGB-level taxonomy.
- New WAAFLE BLAST database and a taxonomy file derived from the chocophlan.v202210_202403 gene family database.
- Improved handling and parsing of tab-delimited files, including how WAAFLE handles gzip files.
- Improved SAM parsing with fixes for edge cases that previously caused failures.
- Optimized performance for processing large datasets.
- Read more in the WAAFLE 1.5 release notes.
- (Option 1) Install the WAAFLE software using PyPi:
$ pip install waafle
- (Option 2) Install the WAAFLE software using conda:
$ conda install waafle -c biobakery- See here for detailed instructions on installing bioBakery conda recipes (e.g. setting channel priorities)
- (Option 3) Install the WAAFLE software manually:
$ git clone https://github.com/biobakery/waafle.git$ python setup.py install- See the WAAFLE manual for other installation options
- Download the WAAFLE BLAST database and taxonomy file:
- Unpack the BLAST database:
$ tar xzfv chocophlan.v202210_202403.tar.gz
conda, as suggested in the quick installation instructions above.- Python 3+ or 2.7+
- Python
numpy(tested with v1.13.3) - NCBI BLAST+ (tested with v2.6.0)
- bowtie2 (for performing read-level QC; tested with v2.2.3)
- You will need a multifasta file containing metagenomic contigs:
- Referred to as
contigs.fnabelow - Or download and try demo_contigs.fna
- Referred to as
- Search your contigs against the WAAFLE database:
-
$ waafle_search contigs.fna chocophlan.v202210_202403.waafledb/chocophlan.v202210_202403.waafledb
- This creates
contigs.blastout(BLAST hits)
-
- Identify ORFs from your contigs and BLAST results:
$ waafle_genecaller contigs.blastout- This creates
contigs.gff(gene calls)
- Taxonomically classify contigs and find LGT events:
$ waafle_orgscorer contigs.fna contigs.blastout contigs.gff chocophlan.v202210_202403.taxonomy.tsv- This creates
contigs.no_lgt.tsv(single-clade contigs) - This creates
contigs.lgt.tsv(putative LGT events)
WAAFLE integrates gene sequence homology and taxonomic provenance to identify metagenomic contigs explained by pairs of microbial clades but not by single clades (i.e. putative LGTs). More specifically, for each locus in a contig, WAAFLE identifies the best hit to each species in a pangenome database. WAAFLE then looks for a species whose minimum per-locus score exceeds a lenient homology threshold (k1). If one or more species meet this criterion, then the contig is assigned to the species with the best average score. Otherwise, the process is repeated for pairs of species. If all per-locus scores for a pair of species exceed a stringent homology threshold (k2), then the contig is considered a putative LGT between those species.
Consider the following pair of examples:
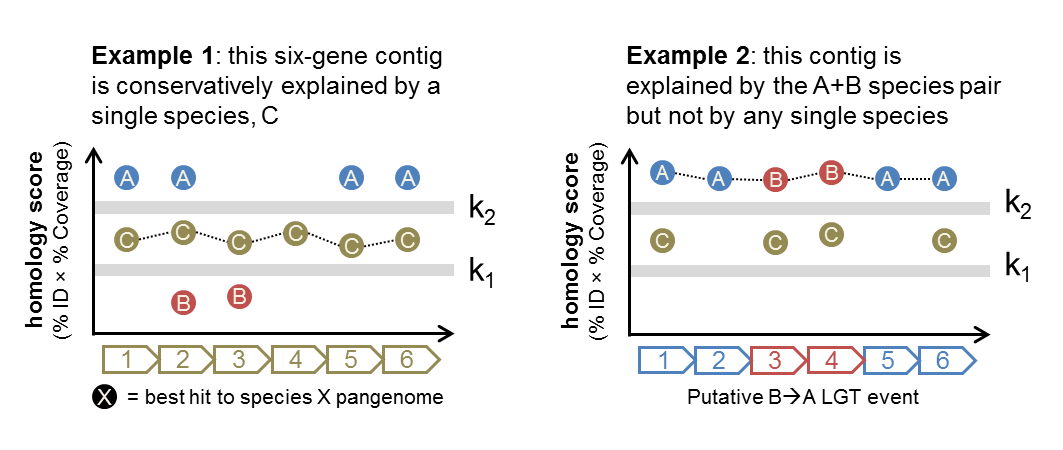
Both cases consider contigs with six protein-coding loci (determined from WAAFLE itself or an independent ORF-calling program such as Prodigal). In Example 1, genes from species C are able to explain all of the loci reasonably well (with scores exceeding k1). Hence, WAAFLE will report this contig as a one-species contig explained by species C.
In Example 2, no single species can explain all of the loci (the minimum score for each species is below k1). However, the pair of species A and B have strong hits (>k2) to all loci, and so WAAFLE concludes that this contig may represent an A+B LGT. Given the AABBAA synteny pattern, a B-to-A transfer would appear to be the more likely mechanism.
Note that in Example 2, if species C had hits to the 2nd and 5th loci that exceeded k1 (as in Example 1), WAAFLE’s algorithm would conservatively favor the weaker one-species explanation for the contig rather than invoking a two-species (LGT-based) explanation.
- WAAFLE 1.0 This version comes with the publication data (DOI: 10.1038/s41564-024-01881-w)