
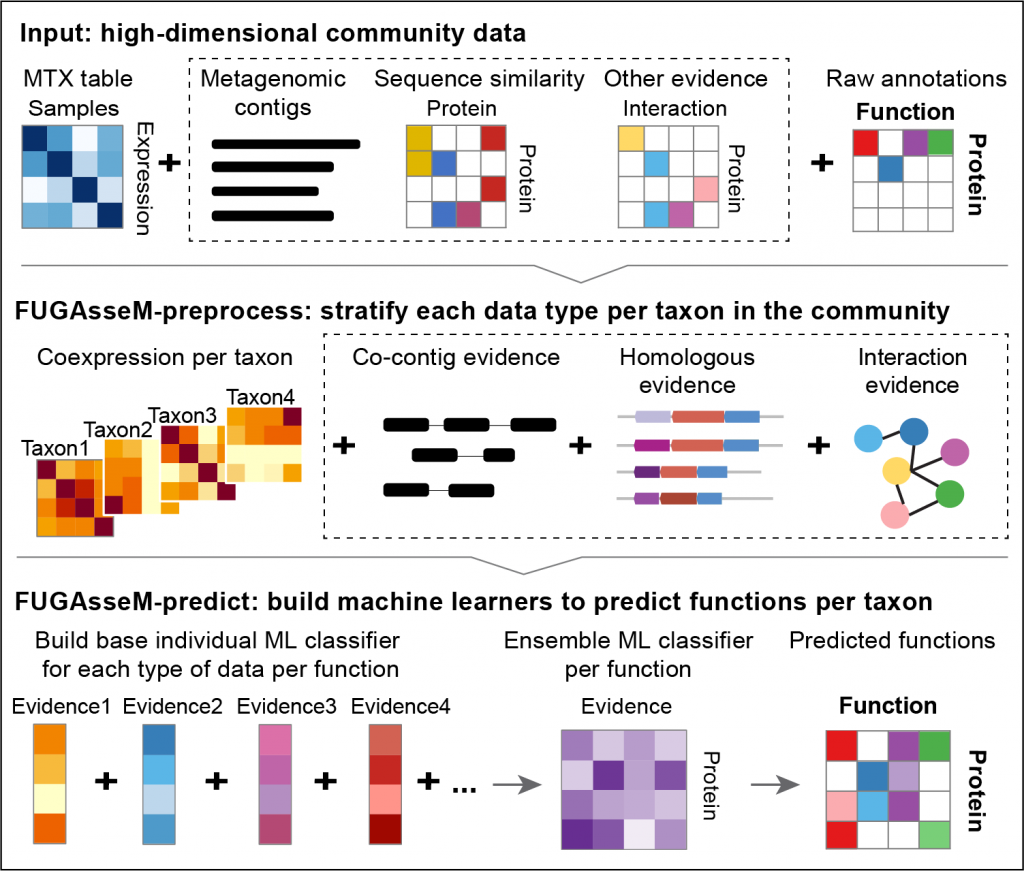
FUGAsseM (Function predictor of Uncharacterized Gene products by Assessing high-dimensional community data in Microbiomes) is a computational tool based on a “guilt by association” approach to predict functions of novel gene products in the context of microbial communities. It uses machine learning methods to predict functions of microbial proteins by integrating multiple types of community-wide data.
User manual || Tutorial || Forum
Citation:
Yancong Zhang, Amrisha Bhosle, Sena Bae, Kelly Eckenrode, Xueying Huang, Jingjing Tang, Danylo Lavrentovich, Lana Awad, Ji Hua, Ya Wang, Xochitl C. Morgan, Bin Li, Andy Krueger, Wendy S. Garrett, Eric A. Franzosa, Curtis Huttenhower. Predicting functions of uncharacterized gene products from microbial communities. Nature Biotechnology (2025)
In the meantime, please add the software link in your Methods if you cite FUGAsseM:
http://huttenhower.sph.harvard.edu/fugassem
For more detailed information about the software, read FUGAsseM User Manual and FUGAsseM Tutorial
FUGAsseM uses a “guilt-by-association” approach by building an individual classifier for upweight individual data type resulting in an evidence weighting behavior in the first layer, followed by a second layer building an ensemble classifier by integrating the weighted learning results from the first layer. This layered learning and predicting process integrates different source of functional information while simultaneously assigning weights to each data type for final predictions.
Requirements
- Python (version >= 3.7, requiring numpy, pandas, multiprocessing, sklearn, matplotlib, scipy, goatools, statistics, tying python packages; tested 3.7, 3.10)
- AnADAMA2 (version >= 0.8.0; tested 0.8.0, 0.10.0)
Install FUGAsseM
You only need to do any one of the following options to install the FUGAsseM package.
Option 1: Installing with conda
- Step1: Add Conda Channels
conda config --add channels defaults
conda config --add channels bioconda
conda config --add channels conda-forge
conda config --add channels biobakery- Step2: Create a conda environment with fugassem installed
$ conda create -n fugassem_env biobakery::fugassem
Option 2: Installing with pip
$ pip install fugassem- If you do not have write permissions to
/usr/lib/, then add the option --user to the install command. This will install the python package into subdirectories of~/.local/. Please note when using the --user install option on some platforms, you might need to add~/.local/bin/to your $PATH as it might not be included by default. You will know if it needs to be added if you see the following messagefugassem: command not foundwhen trying to run FUGAsseM after installing with the --user option.
A typical process runs FUGAsseM per dataset.
For a list of command line options, run:$ fugassem --helpThis command yields:
usage: fugassem_workflow.py [-h] [--version]
[--taxon-level {MSP,Terminal,Species,Genus,Family,
Order,Class,Phylum}]
...Run the canonical function prediction of FUGAsseM, which requires MTX abundance data and annotation of protein families for function prediction.
Input files:
- Normalized MTX abundance table stratified by taxa, e.g. demo_proteinfamilies_rna_CPM.stratified_Species_mtx.tsv
- GO (raw) annotations for protein families, e.g. demo_proteinfamilies.GO.simple.tsv
Demo run of FUGAsseM-MTX model
$ fugassem --basename $BASENAME \
--input $INPUT_MTX \
--input-annotation $INPUT_annotation \
--output $OUTPUT_DIR-
- $INPUT_MTX = the protein families MTX abundances file (TSV format)
- $INPUT_annotation = raw GO annotations for some of these protein families (TSV format)
- $OUTPUT_DIR = the output folder
- Output files will be created named with $BASENAME:
$OUTPUT_DIR/merged/$BASENAME.finalized_ML.prediction.tsv: this file combines the finalized predictions from all taxa by using machine learning approaches based on MTX coexpression patterns (TSV format).- Predictions files of each taxon will also be created. E.g. the finalized predictions using MTX-coexpression evidence per taxon are in the file:
$OUTPUT_DIR/main/$TAXON_NAME/prediction/finalized/$BASENAME.$TAXON_NAME.finalized_ML.prediction.tsv
- Output files will be created named with $BASENAME:
- Run the integrated function prediction workflow of FUGAsseM. When other community-wide data are available, FUGAsseM can predict functions by integrating multiple pieces of evidence. The additional steps in this workflow are 1) building individual machine learning classifiers for each type of evidence including coexpression as discussed above, 2) and integration to generate an ensemble classifier for final function prediction.
- Input files
- normalized MTX-based abundance table stratified by taxa (TSV format file), e.g. demo_proteinfamilies_rna_CPM.stratified_Species_mtx.tsv
- raw annotations for protein families (TSV format file), e.g. demo_proteinfamilies.GO.simple.tsv
- vector-based evidence file (TSV format file), e.g. demo_proteinfamilies.GO.homology.tsv
- matrix-based evidence files (TSV format file), e.g.
- demo_proteinfamilies.DDI.simple.tsv: Domain-Domain interactions for building DDI network for prediction
- demo_proteinfamilies.contig.simple.tsv: TAXON_NAMEMGX-based contigs of protein families for building co-contig network for prediction
- Input files
Demo run of FUGAsseM-full model
$ fugassem --basename $BASENAME \
--input $INPUT_MTX \
--input-annotation $INPUT_annotation \
--vector-list $VECTOR_list --matrix-list $METRIX_list \
--output $OUTPUT_DIR
- $INPUT_MTX = the protein families MTX abundances file (TSV format)
- $INPUT_annotation = raw GO annotations for some of these protein families (TSV format)
- $VECTOR_list = file names of vector-based evidence data, provided as a string of 'file1,file2', semi-colon delimited for multiple files.
- $METRIX_list = file names of matrix-based evidence data, provided as a string of 'file1,file2', semi-colon delimited for multiple files.
- $OUTPUT_DIR = the output folder
- Output files will be created named with $BASENAME:
$OUTPUT_DIR/merged/$BASENAME.finalized_ML.prediction.tsv: this file combines the finalized predictions from all taxa by using machine learning approaches based on MTX coexpression patterns (TSV format).$OUTPUT_DIR/merged/$BASENAME.$EVIDENCE_TYPE_ML.prediction.tsv(where$EVIDENCE_TYPE= the basename of each piece of evidence): this file includes combined predictions based on individual type of evidence (TSV format file).- Predictions files of each taxon will also be created:
- FUGAsseM predicts functions based on input evidence data.
- The finalized prediction results using integrated evidence per taxon are in the file:
$OUTPUT_DIR/main/$TAXON_NAME/prediction/finalized/$BASENAME.$TAXON_NAME.finalized_ML.prediction.tsv. - The prediction results by using individual evidence per taxon are in the file:
$OUTPUT_DIR/$TAXON_NAME/prediction/$EVIDENCE_TYPE/$BASENAME.$TAXON_NAME.$EVIDENCE_TYPE_ML.prediction.tsv(where$EVIDENCE_TYPE= the basename of each piece of evidence).
-
Download pre-computed predicted annotations by FUGAsseM for microbial communities. For more information, see our paper "Predicting functions of uncharacterized gene products from microbial communities", Nature Biotechnology (2025)
We provide FUGAsseM's predicted functions of the protein families assembled from the Integrative Human Microbiome Project (HMP2), Inflammatory Bowel Disease Multi'omics Database (IBDMDB). Here, predicted functions cover all categories of the Gene Ontology (i.e. Biological Process, Molecular Function and Cellular Component).
- HMP2_fugassem_GO.func_assignment.tsv.gz (26 MB): high-confidence GO annotations for HMP2 protein families predicted by FUGAsseM
- HMP2_fugassem_GO.func_assignment.raw.tsv.gz (2.4 GB): raw GO annotations for HMP2 protein families predicted by FUGAsseM
- HMP2_proteinfamilies_mtx.raw_genes_all.final.tsv.gz (11 MB): HMP2 protein families detected by the MTX used in this study
- HMP2_fugassem_GO.func_assignment.protein_sequences.fasta.gz (108 MB): protein sequences of HMP2 protein families processed by FUGAsseM in this study
- HMP2_fugassem_GO.func_assignment.protein_sequences.NH.fasta.gz (752 K): protein sequences of HMP2 No-Homology (NH) protein families processed by FUGAsseM in this study
- For more information about the HMP2 protein families generated by MetaWIBELE and their associated publication, visit MetaWIBELE summary page.