
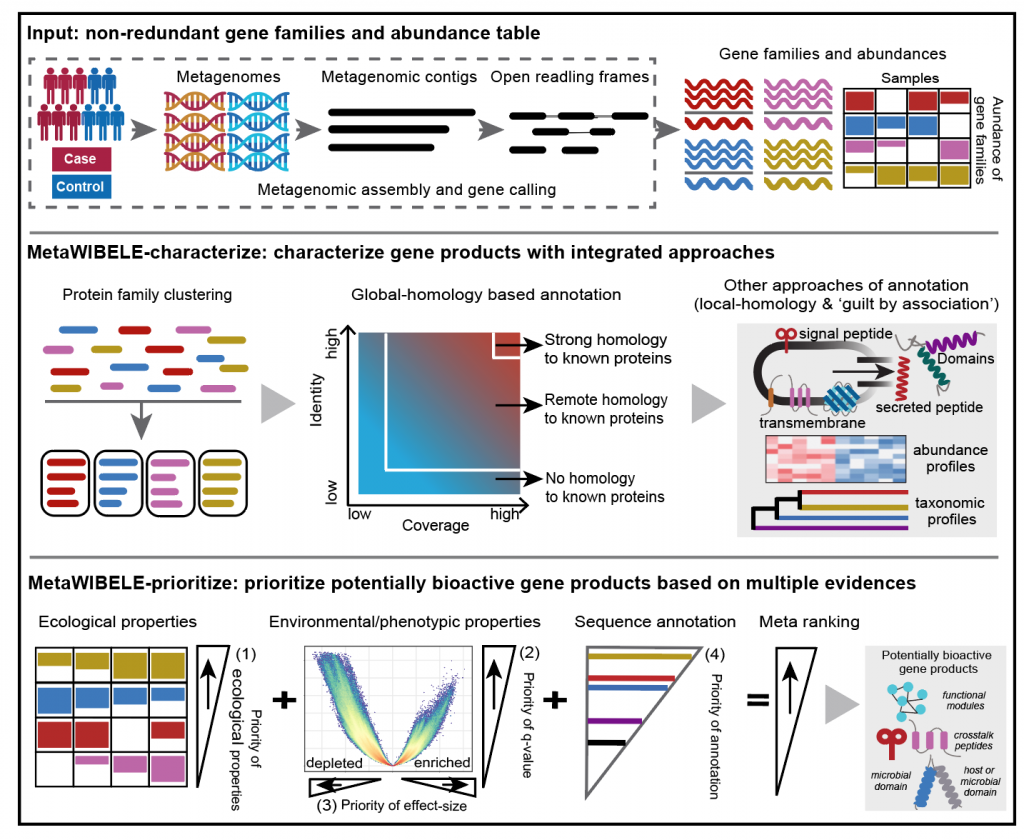
MetaWIBELE (Workflow to Identify novel Bioactive Elements in the microbiome) is a workflow to efficiently and systematically identify and prioritize potentially bioactive (and often uncharacterized) gene products in microbial communities. It prioritizes candidate gene products from assembled metagenomes using a combination of sequence homology, secondary-structure-based functional annotations, phylogenetic binning, ecological distribution, and association with environmental parameters or phenotypes to target candidate bioactives.
User manual || Tutorial || Forum
Citation:
Yancong Zhang, Amrisha Bhosle, Sena Bae, Lauren J. McIver, Gleb Pishchany, Emma K. Accorsi, Kelsey N. Thompson, Cesar Arze, Ya Wang, Ayshwarya Subramanian, Sean M. Kearney, April Pawluk, Damian R. Plichita, Gholamali Rahnavard, Afrah Shafquat, Ramnik J. Xavier, Hera Vlamakis, Wendy S. Garrett, Andy Krueger, Curtis Huttenhower*, Eric A. Franzosa*. Discovery of bioactive microbial gene products in inflammatory bowel disease. Nature, 606: 754–760 (2022)
And feel free to link to MetaWIBELE in your Methods: http://huttenhower.sph.harvard.edu/metawibele
For more detailed information about the software, read MetaWIBELE User Manual and MetaWIBELE Tutorial
Requirements
- Python (version >= 3.6, requiring numpy, pandas, scipy packages; tested 3.6, 3.7)
- AnADAMA2 (version >= 0.7.4; tested 0.7.4, 0.8.0)
- CD-hit (version >= 4.7; tested 4.7)
- Diamond (version >= 0.9.24; tested 0.9.24)
- MSPminer (version >= 1.0.0; licensed software; tested version 1.0.0)
- MaAsLin2 (version >= 1.5.1; tested 1.5.1)
- Interproscan (version >= 5.31-70) (installing with activating Phobius/SignalP/TMHMM analyses; InterProScan 5.51-85.0 or later are recommended for potential simpler installation; tested 5.31-70, 5.51-85.0; you can skip to install Interproscan if you’d like to ignore domain/motif annotation by running MetaWIBELE with “--bypass-interproscan”)
- Signalp (version >= 4.1; licensed software; installing with integrating in interproscan; used for domain/motif annotation; tested 4.1)
- TMHMM (version >= 2.0; licensed software; installing with integrating in interproscan; used for domain/motif annotation; tested 2.0)
- Phobius (version >= 1.01; licensed software; installing with integrating in interproscan; used for domain/motif annotation; tested 1.01)
- PSORTb (version >= 3.0) (licensed software; used for domain/motif annotation; tested 3.0; you can skip to install psortb if you’d like to ignore protein subcellular localization annotations by running MetaWIBELE with “--bypass-psortb”)
- Optional: only required if using MetaWIBELE utilities to prepare inputs for MetaWIBELE using metagenomic sequencing reads
- MEGAHIT (version >= 1.1.3; tested 1.1.3)
- Prokka (version >= 1.14-dev; recommend to not set '-c' parameter when running prodigal with metagenome mode; tested 1.14-dev)
- Prodigal (version >= 2.6; tested 2.6)
- SeqKit (version >= 2.6.1; tested 2.6.1)
- Bowtie2 (version >= 2.3.2; tested 2.3.2)
- SAMtools (version >= 1.9; tested 1.9)
- featureCounts (version >= 1.6.2; tested 1.6.2)
Note: Please install the required software in a location in your $PATH. If you always run with gene families (non-redundant gene catalogs), the optional softwares are not required. Also if you always run with one or more bypass options (for information on bypass options, see optional arguments to the section Workflow by bypass mode). The software required for the steps you bypass does not need to be installed.
Install MetaWIBELE
You only need to do any one of the following options to install the MetaWIBELE package.
Option 1: Installing with docker
$ docker pull biobakery/metawibele- This docker image includes most of the dependent software packages.
- Large software packages and those with licenses are NOT included in this image and needed to be installed additionally:
- Users should review the license terms and install these packages manually.
- Softwares with the license : MSPminer, Signalp, TMHMM, Phobius, PSORTb
- Softwares with large size: Interproscan (Note: We recommen dinstalling InterProScan 5.51-85.0 (requiring at least Java 11) or later for potential simpler installation, and active Phobius/SignalP/TMHMM analyses by customizing your
interproscan.propertiesconfiguration, see more details from InterProScan document).
Option 2: Installing with pip
$ pip install metawibele- If you do not have write permissions to
/usr/lib/, then add the option --user to the install command. This will install the python package into subdirectories of~/.local/. Please note when using the --user install option on some platforms, you might need to add~/.local/bin/to your $PATH as it might not be included by default. You will know if it needs to be added if you see the following messagemetawibele: command not foundwhen trying to run MetaWIBELE after installing with the --user option.
Option 3: Installing with conda
$ conda install -c biobakery metawibele
Install databases
To run metawibele, you are required to install the dependent UniRef database. We have built the database based on UniProt/UniRef90 2019_01 sequences and annotations. You can download and uncompress this database (including both sequences and annotations) and provide $UNIREF_LOCATION as the location to install the database.
NOTE: Please point to this location for the default uniref database in the global config file (e.g., "metawibele.cfg", see details in the following section - Prepare configuration file). Alternatively, you can either set the location with the environment variable $UNIREF_LOCATION, or move the downloaded database to the folder named "uniref_database" in the current working directory.
Option 1: Download and uncompress them into $UNIREF_LOCATION as the location to install the database:
- UniRef90 sequences indexed by Diamond v0.9.24 (20.0GB): uniref90.fasta.dmnd.tar.gz
- UniRef90 annotations (2.8 GB): uniref90_annotations.tar.gz
Option 2: Run the following commands to install the database into the location $UNIREF_LOCATION:
- Download UniRef90 sequences indexed by Diamond v0.9.24 into $UNIREF_LOCATION:
$ metawibele_download_database --database uniref --build uniref90_diamond --install-location $UNIREF_LOCATION
* Download UniRef90 annotations into $UNIREF_LOCATION:
$ metawibele_download_database --database uniref --build uniref90_annotation --install-location $UNIREF_LOCATION
Check install
To check out the install of MetaWIBELE packages and all dependencies (tools and databases), run the command:
$ metawibele_check_install- By default, it will check both MetaWIBEKE packages and all dependencies.
- Alternatively, add the option "--types {metawibele,required,optional,all}" to check the install of specific package or dependency.
Prepare configuration file
To run MetaWIBELE, you are required to customize the global configuration file "metawibele.cfg" and make sure that it's in your current working directory. You can use one of the following options to get the configuration template.
NOTE: De default, MetaWIBELE will use the global configurations from "metawibele.cfg" in the current working directory. Alternatively, you can always provide the location of the global configuration file you would like to use with the "--global-config " option to metawibele (see more in the following section - How to run).
- Download "metawibele.cfg" into your current working directory by any one of the following options:
- Option 1: Obtain copies by right-clicking the link and selecting "save link as": metawibele.cfg
- Option 2: Run this command to download the global configuration file:
$ metawibele_download_config --config-type global
- Customize your configurations in "metawibele.cfg" before running MetaWIBELE:
- Customize the path of dependent databases (required):
[database] # The absolute path of uniref databases folder. uniref_db = # The domain databases used by MetaWIBELE. [data_path] provide the absolute path of the domain databases folder; [none] use the default domain databases installed in the metawibele package. [ Default: none ] domain_db = none
A typical process runs MetaWIBELE-characterize workflow and then MetaWIBELE-prioritize workflow per dataset.
- For a list of all available workflows, run:
$ metawibele --help
This command yields:
usage: metawibele [-h] [--global-config GLOBAL_CONFIG]
{characterize,prioritize,preprocess}
MetaWIBELE workflows: A collection of AnADAMA2 workflows
positional arguments:
{profile,characterize,prioritize,preprocess}
workflow to run
optional arguments:
-h, --help show this help message and exit
--global-config GLOBAL_CONFIG the global configuration file of MetaWIBELE (default: None)NOTE: De default, MetaWIBELE will use the global configurations from “metawibele.cfg” in the current working directory. Alternatively, you can always provide the location of the global configuration file you would like to use with the "--global-config " option to metawibele.
- All workflows follow the general command format:
$ metawibele $WORKFLOW
- For specific options of workflow, run:
$ metawibele $WORKFLOW --help
- For example:
$ metawibele characterize --help
This command yields:
usage: characterize.py [-h] [--version] [--threads THREADS]
[--characterization-config CHARACTERIZATION_CONFIG]
[--mspminer-config MSPMINER_CONFIG]
[--bypass-clustering] [--bypass-global-homology]
[--bypass-domain-motif] [--bypass-interproscan]
[--bypass-pfamtogo] [--bypass-domine] [--bypass-sifts]
[--bypass-expatlas] [--bypass-psortb]
[--bypass-abundance] [--bypass-mspminer] [--bypass-maaslin] [--split-number SPLIT_NUMBER]
[--bypass-integration] [--study STUDY]
[--basename BASENAME] --input-sequence INPUT_SEQUENCE
--input-count INPUT_COUNT
[--input-metadata INPUT_METADATA] [--output OUTPUT]
[-i INPUT] [--config CONFIG] [--local-jobs JOBS]
[--grid-jobs GRID_JOBS] [--grid GRID]
[--grid-partition GRID_PARTITION]
[--grid-benchmark {on,off}]
[--grid-options GRID_OPTIONS]
[--grid-environment GRID_ENVIRONMENT]
[--grid-scratch GRID_SCRATCH] [--dry-run]
[--skip-nothing] [--quit-early]
[--until-task UNTIL_TASK] [--exclude-task EXCLUDE_TASK]
[--target TARGET] [--exclude-target EXCLUDE_TARGET]
[--log-level {DEBUG,INFO,WARNING,ERROR,CRITICAL}]
A workflow for MetaWIBELE characterization- Run MetaWIBELE-characterize workflow, which uses gene families (non-redundant gene catalogs) to build protein families and annotate them functionally and taxonomically.
- Input files for for characterization
- protein sequences for non-redundant gene families (Fasta format file), e.g. demo_genecatalogs.centroid.faa
- reads counts table for non-redundant gene families (TSV format file), e.g. demo_genecatalogs_counts.all.tsv
- metadata file (TSV format file), e.g. demo_mgx_metadata.tsv
- the global configuration file in the current working directory, e.g. metawibele.cfg
- Demo run of MetaWIBELE-characterize
$ metawibele characterize --input-sequence $INPUT_SEQUENCE --input-count $INPUT_COUNT --input-metadata $INPUT_METADATA --output $OUTPUT_DIR- Make sure the global configuration file "metawibele.cfg" is in your working directory.
- $INPUT_SEQUENCE = the protein sequences file for gene families (non-redundant gene catalogs)
- $INPUT_COUNT = the count file for gene families (non-redundant gene catalog)
- $INPUT_METADATA = the metadata file
- $OUTPUT_DIR = the output folder
- Four main output files will be created where $BASENAME is the basename of output files provided in "metawibele.cfg":
- $OUTPUT_DIR/finalized/$BASENAME_proteinfamilies_annotation.tsv
- $OUTPUT_DIR/finalized/$BASENAME_proteinfamilies_annotation.attribute.tsv
- $OUTPUT_DIR/finalized/$BASENAME_proteinfamilies_annotation.taxonomy.tsv
- $OUTPUT_DIR/finalized/$BASENAME_proteinfamilies_nrm.tsv
- Intermediate temp files will also be created:
- $OUTPUT_DIR/clustering
- a subfolder including the full outputs from clustering proteins into proteins family
- $OUTPUT_DIR/global_homology_annotation
- a subfolder including the full outputs from global-homology based annotation
- $OUTPUT_DIR/domain_motif_annotation
- a subfolder including the full outputs from domain-motif based annotation
- $OUTPUT_DIR/abundance_annotation
- a subfolder including the full outputs from abundance-based annotation
- $OUTPUT_DIR/clustering
- Input files for for characterization
- Run MetaWIBELE-prioritize workflow, which ranks protein families (with a focus on uncharacterized, and thus particularly novel, families) by combining evidence from sample-specific feature ecology and host disease phenotypes or environmental parameters.
- Input files for prioritization
- annotation file produced by MetaWIBELE-characterize workflow (TSV format file), e.g. demo_proteinfamilies_annotation.tsv
- annotation attribute file produced by MetaWIBELE-characterize workflow (TSV format file), e.g. demo_proteinfamilies_annotation.attribute.tsv
- Demo run of MetaWIBELE-prioritize
$ metawibele prioritize --input-annotation $INPUT_ANNOTATION --input-attribute $INPUT_ATTRIBUTE --output $OUTPUT_DIR- Make sure the global configuration file "metawibele.cfg" is in your working directory.
- $INPUT_ANNOTATION = the final annotation file produced by MetaWIBELE-characterize workflow
- $INPUT_ATTRIBUTE = the final attribute file produced by MetaWIBELE-characterize workflow
- $OUTPUT_DIR = the output folder
- Three main output files will be created where $BASENAME is the basename of output files provided in "metawibele.cfg":
- $OUTPUT_DIR/$BASENAME_unsupervised_prioritization.rank.table.tsv
- $OUTPUT_DIR/$BASENAME_supervised_prioritization.rank.table.tsv
- $OUTPUT_DIR/$BASENAME_supervised_prioritization.rank.selected.table.tsv
- Input files for prioritization
- Parallelization Options
When running any workflow you can add the following command-line options to make use of existing computing resources:- --local-jobs <1> : Run multiple tasks locally in parallel. Provide the max number of tasks to run at once. The default is one task running at a time.
- --grid-jobs <0> : Run multiple tasks on a grid in parallel. Provide the max number of grid jobs to run at once. The default is zero tasks are submitted to a grid resulting in all tasks running locally.
- --grid <slurm> : Set the grid available on your machine. This will default to the grid found on the machine with options of slurm and sge.
- --grid-partition <serial_requeue> : Jobs will be submitted to the partition selected. The default partition selected is based on the default grid.
- For additional workflow options, see the AnADAMA2 user manual.
Download pre-computed prioritizations and characterization annotations of MetaWIBELE for different microbial communities. For more information, see our paper "Discovery of bioactive microbial gene products in inflammatory bowel disease", Nature, 606: 754–760 (2022)
Applying MetaWIBELE to 1,595 HMP2 metagenomes
These are prioritizations and characterization annotations for assemblies and gene families from the Integrative Human Microbiome Project (HMP2), Inflammatory Bowel Disease Multi'omics Database (IBDMDB). Prioritization indicates predicted bioactivity in the human gut during inflammatory bowel disease.
- Gene families assembled
- HMP2_contig_sequence.fasta.gz (26 GB): metagenomic contig sequences
- HMP2_gene_info.tsv.gz (2.3 GB): information of open reading frames
- HMP2_genefamilies.clstr.gz (279 MB): clustering information for gene families (non-redundant gene catalogs)
- HMP2_genefamilies.centroid.fna.gz (554 MB): nucleotide sequences of representatives for gene families
- HMP2_genefamilies.centroid.faa.gz (335 MB): protein sequences of representatives of gene families
- HMP2_genefamilies_counts.tsv.gz (699 MB): reads counts of gene families
- Characterization of protein families
- HMP2_proteinfamilies_annotation.tsv.gz (675 MB): main annotations of protein families
- HMP2_proteinfamilies_annotation.attribute.tsv.gz (2.8 GB): attributes of annotation types
- HMP2_proteinfamilies_annotation.taxonomy.tsv.gz (96 MB): relative abundance of protein families
- HMP2_proteinfamilies_nrm.tsv.gz (1.5 GB): normalized DNA abundance of protein families
- HMP2_proteinfamilies_rna_nrm.tsv.gz (287 MB): normalized RNA abundance of protein families
- HMP2_proteinfamilies.clstr.gz (29 MB): clustering information for protein families
- HMP2_proteinfamilies.centroid.faa.gz (270 MB): protein sequences of centroids for protein families
- Prioritization of protein families
- HMP2_unsupervised_prioritization.rank.table.tsv.gz (76 MB): prioritization based on ecological properties
- HMP2_supervised_prioritization.rank.table.tsv.gz (134 MB): prioritization based on ecological and phenotypic properties
Applying MetaWIBELE to 45 Red Sea metagenomes
These are prioritizations and characterization annotations for assemblies and gene families from previously published Red Sea metagenomes (PMID: 27377622). Prioritization indicates predicted bioactivity in the Red Sea during adaptation to a wider range of niches in harsher deep, cold, dark, and salty waters.
- Gene families assembled
- RedSea_contig_sequence.fasta.gz (748 MB): metagenomic contig sequences
- RedSea_gene_info.tsv.gz (79 MB): information of open reading frames
- RedSea_genefamilies.clstr.gz (13 MB): clustering information for gene families (non-redundant gene catalogs)
- RedSea_genefamilies.centroid.fna.gz (118 MB): nucleotide sequences of representatives for gene families
- RedSea_genefamilies.centroid.faa.gz (70 MB): protein sequences of representatives of gene families
- RedSea_genefamilies_counts.tsv.gz (17 MB): reads counts of gene families
- Characterization of protein families
- RedSea_proteinfamilies_annotation.tsv.gz (113 MB): main annotations of protein families
- RedSea_proteinfamilies_annotation.attribute.tsv.gz (641 MB): attributes of annotation types
- RedSea_proteinfamilies_annotation.taxonomy.tsv.gz (17 MB): relative abundance of protein families
- RedSea_proteinfamilies_nrm.tsv.gz (34 MB): normalized DNA abundance of protein families
- RedSea_proteinfamilies.clstr.gz (6.7 MB): clustering information for protein families
- RedSea_proteinfamilies.centroid.faa.gz (63 MB): protein sequences of centroids for protein families
- Prioritization of protein families
- RedSea_unsupervised_prioritization.rank.table.tsv.gz (23 MB): prioritization based on ecological properties
- RedSea_supervised_prioritization.rank.table.tsv.gz (49 MB): prioritization based on ecological and phenotypic properties